Name:
Tell:
Email:
Organization:
Peach Transcriptome Research Revealed the Complexity of Fruit Tree Functional Genomics
2013-07-10
Peach (Prunus persica), a member of the family Rosaceae, is the third most important deciduous fruit trees worldwide, ranking only after apple and pear. Peach fresh, juice and cans are well received by consumers. With the increasing demand for peach and related products, it is urgent to find more effective way for new varieties breeding. Therefore, the functional genomics studies of peach will have great power on promoting the peach breeding and industry.
To facilitate gene isolation which controlling important horticultural traits of peach, Dr. WANG Lu from Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Wuhan Botanical Garden conducted transcriptome sequencing. A total of 133 million pair-end RNA-Seq reads were generated from leaf, flower, and fruit, and 90% of reads were mapped to the peach draft genome. Sequence annotation revealed 1,162 transcription factors and 2,140 novel transcribed regions (NTRs). Of these 2,140 NTRs, 723 contain an open reading frame, while the rest 1,417 are non-coding RNAs. A total of 9,587 SNPs were identified across six peach genotypes, with an average density of one SNP per ~5.7kb. The top of chromosome 2 has higher density of expressed SNPs than the rest of the peach genome. The average density of SSR is 312.5/Mb, with tri-nucleotide repeats being the most abundant. Most of the detected SSRs are AT-rich repeats and the most common di-nucleotide repeat is CT/TC. The predominant type of alternative splicing (AS) events in peach is exon-skipping isoforms, which account for 43% of all the observed AS events. In addition, the most active transcribed regions in peach genome were also analyzed.
Their study reveals for the first time the complexity of the peach transcriptome, and gives an extensive new knowledge about alternative splicing, NTRs, and gene boundaries. The results will not only serve as a complement to the predicted gene database of peach, but also provide an invaluable resource for functional genomics research in peach and other fruit trees in the future.
This project was supported by funds received from the National 863 program of China (No. 2011AA100206), the National 948 Project from the Ministry of Agriculture of China, and the National Natural Science Foundation of China (No. 31201604 and 31000139). Results were published in Plant Molecular Biology entitled "Deep RNA-Seq uncovers the peach transcriptome landscape”.
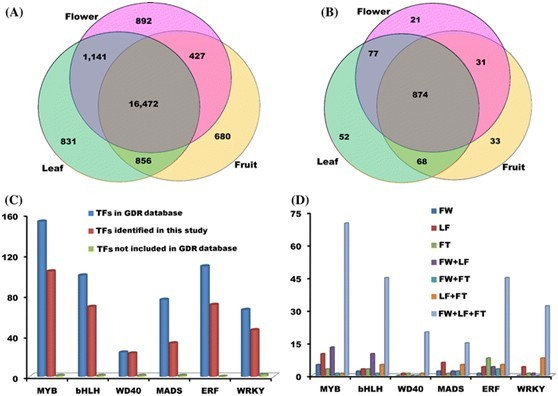
Characterization of genes in the peach transcriptome (Image by Dr. WANG)

