Name:Pingfang Yang
Tell:
Email:yangpf@hubu.edu.cn
Organization: Wuhan Botanical Garden
Researchers Apply Full-length Transcriptome Sequencing Technology in Lotus
2019-07-10
Alternative splicing (AS) events play a critical role in regulating different physiological and developmental processes in eukaryotes by dramatically increasing the diversity of the transcriptome and proteome. However, due to the read length limitation of next-generation sequencing technique, the AS could not be comprehensively identified and deeply investigated in lotus (Nelumbo nucifera).
To get a comprehensive survey of AS survey in lotus, ZHANG Yue from Wuhan Botanical Garden, supervised by Prof. YANG Pingfang, pooled a total of 19 different samples and applied both PacBio single-molecule read-time (SMRT) sequencing and Illumina -based sequencing of the transcriptome from 16 different tissues of ‘China Antique’.
By using bioinformatics methods, a total of 104,288 AS events from 16,543 genes were identified, with alternative 3’ splice-site being the predominant model, following by intron retention. Moreover, 370 AS events were tissue-specific expressed among 12 tissues. Additionally, the identified tissue-specific genes and isoforms were found to played important role in development of corresponding tissue.
The result showed that the annotation of lotus was well updated and the complexity of AS events was comprehensively unveiled. The fusion transcripts and tissue-specific expressed genes were identified by bioinformatics analysis for the first time in lotus.
A global view of gene regulation in lotus is obtained, thus benefiting gene functional studies and molecular breeding of this economically important aquatic plant.
This study was supported by National Natural Science Foundation of China, and results were published in the DNA Research entitled “The complexity of alternative splicing and landscape of tissue-specific expression in lotus (Nelumbo nucifera) unveiled by Illumina- and single-molecule real-time-based RNA-sequencing”
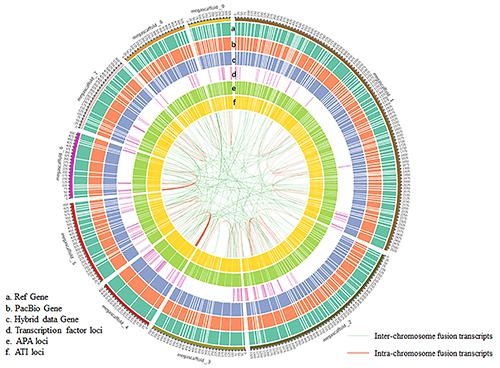
CIRCOS visualization of distribution of different data at genome-wide level. (a) Gene density of reference genome. (b) Density of the predicted genes from PacBio data. (Image by ZHANG Yue)

